🧬 Doing Biology in Julia 🧬
CAJUN
2024-04-24
Packages and ecosystem
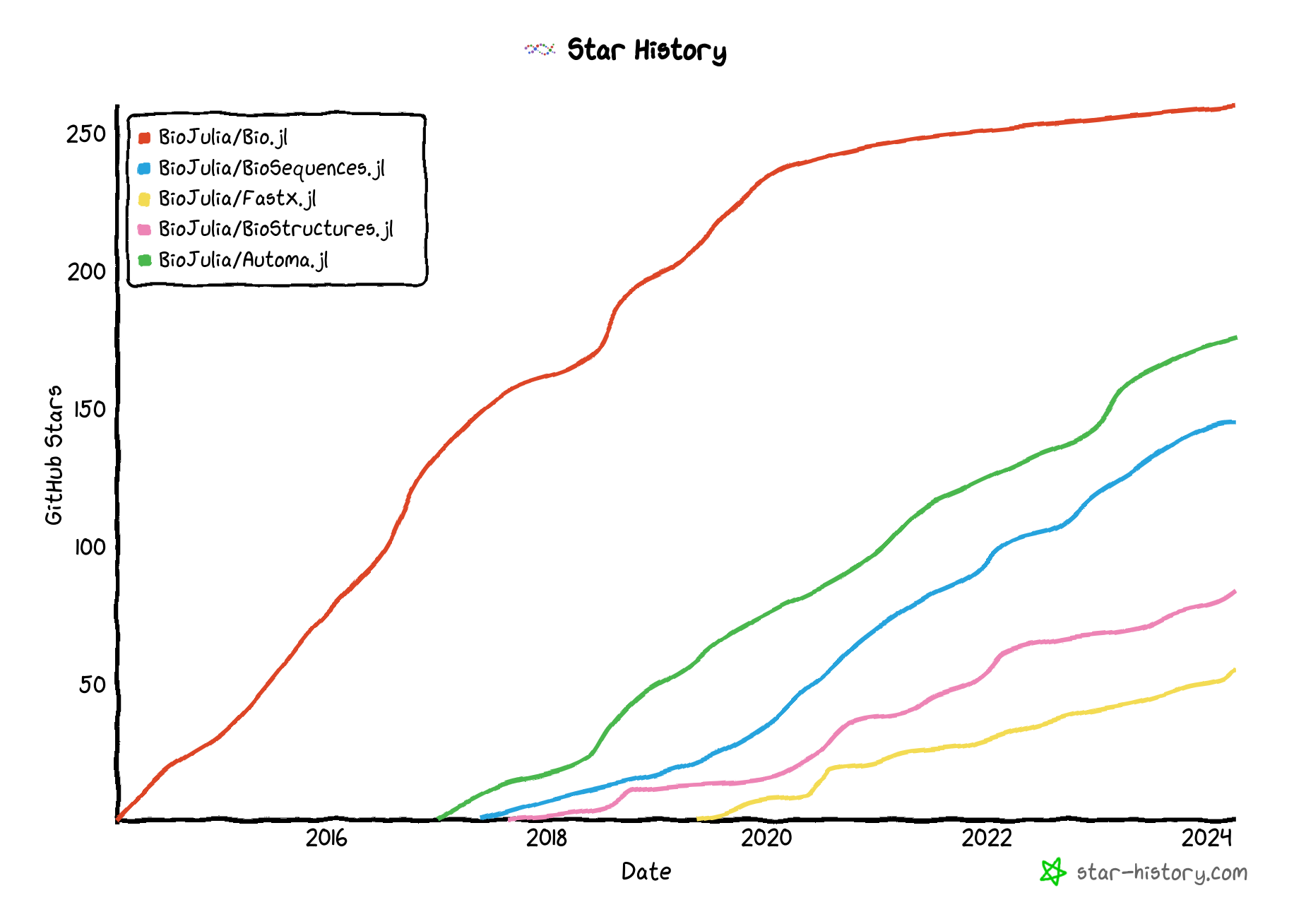
Non-BioJulia bio stuff in julia
Non-BioJulia bio stuff in julia

The central dogma of molecular biology
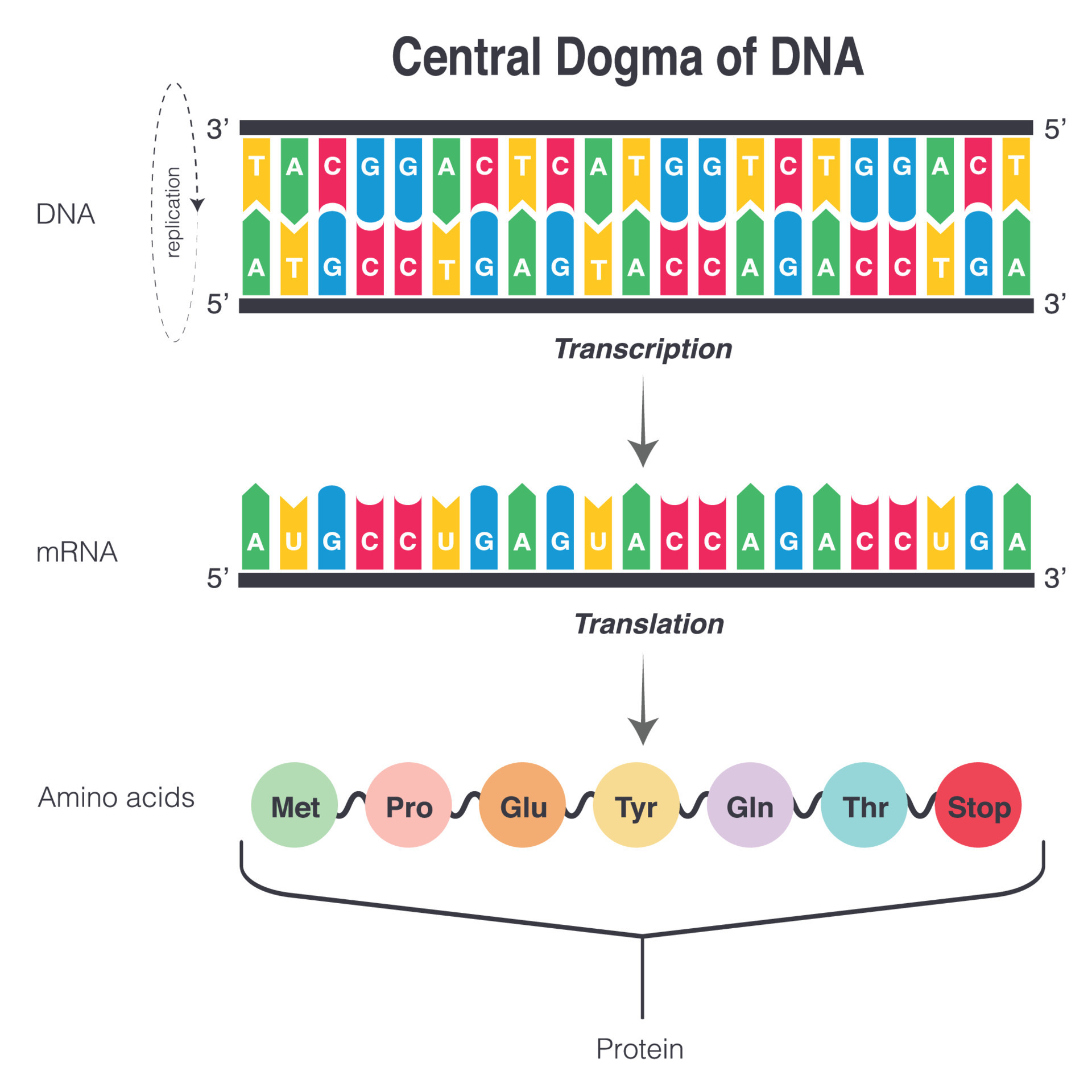
- DNA copied into DNA through “replication”
- DNA read into RNA through “transcription”
- RNA read into protein through “translation”
Major biological molecules are 1D arrays (polymers)
DNA
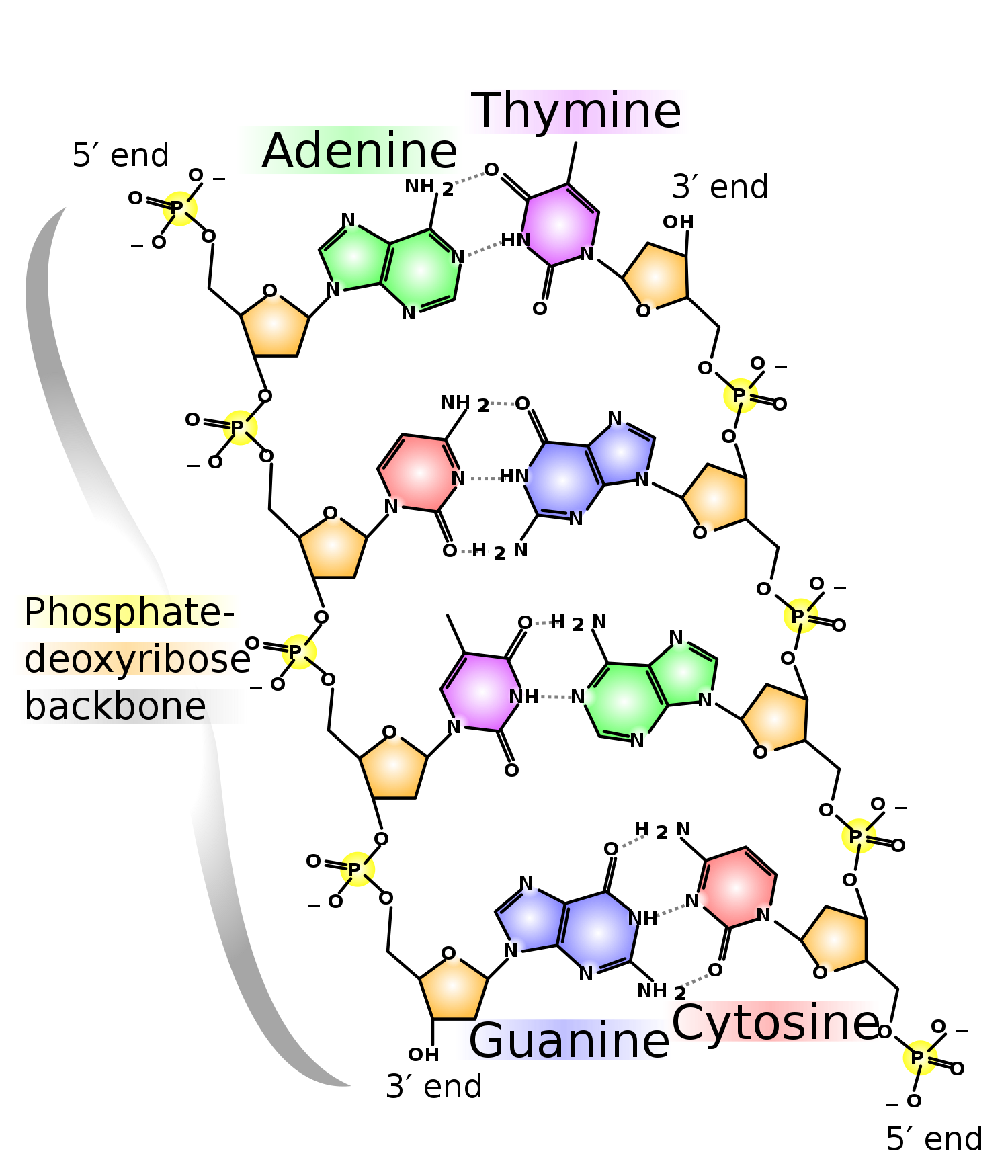
Protein
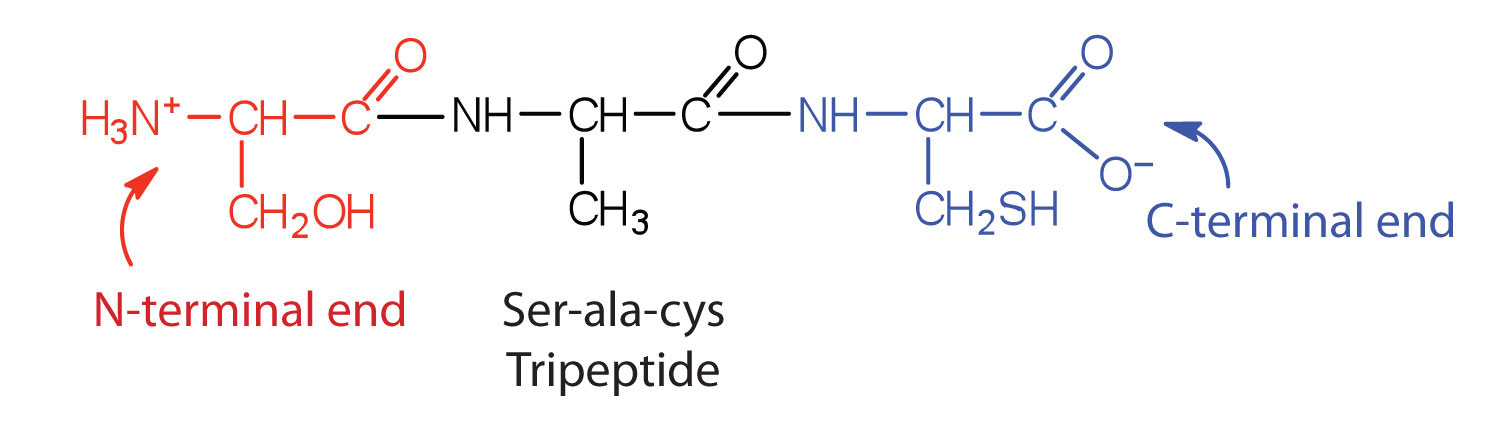
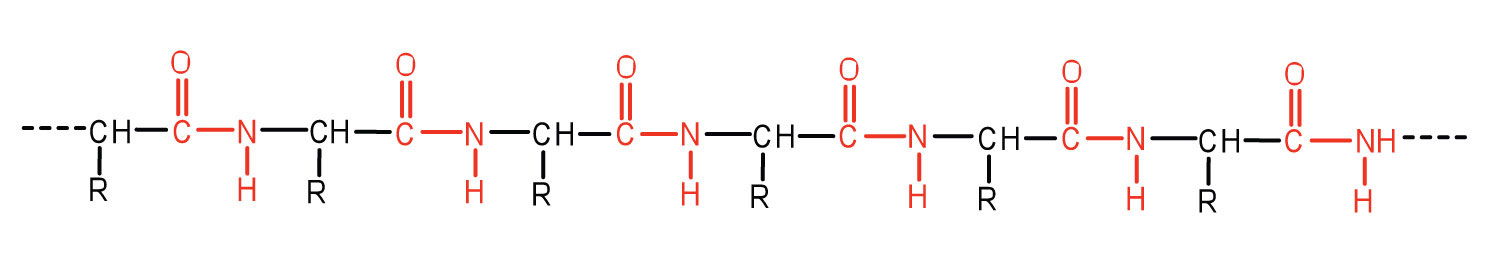
The functions of biomolecules are determined by their sequence
![]()
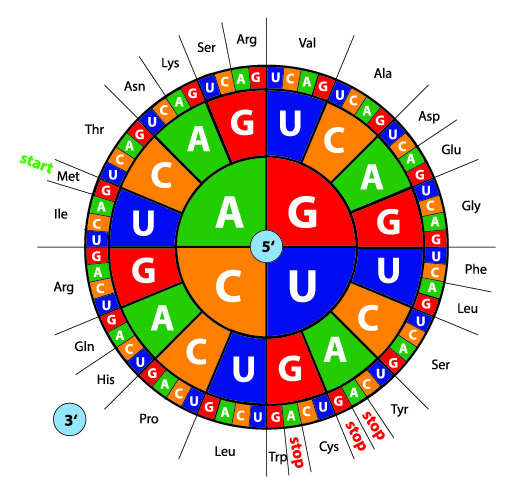
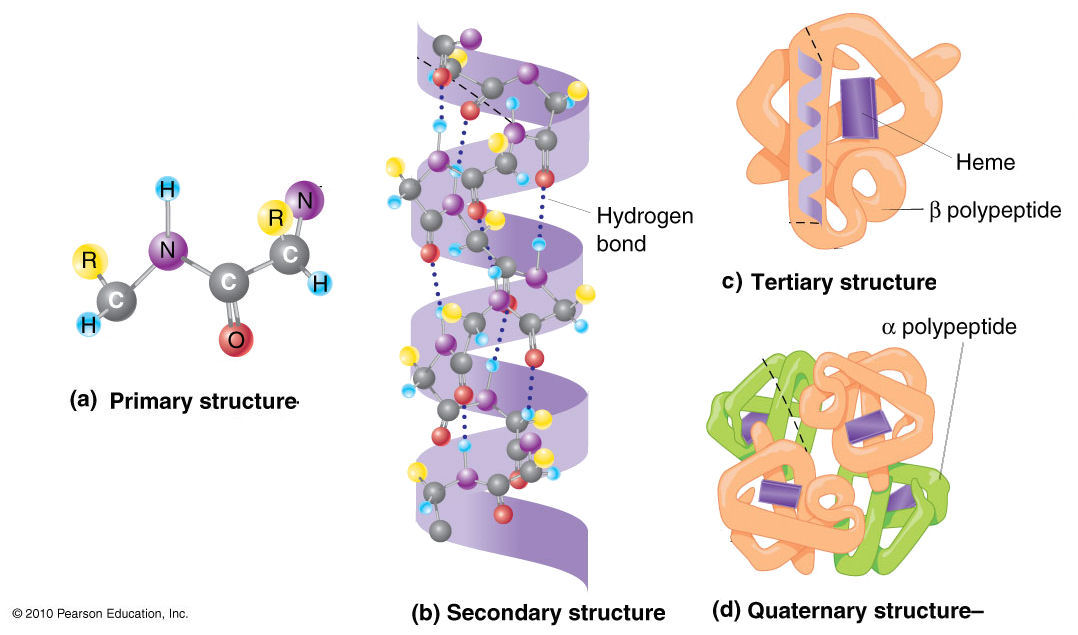
Evolutionary relationships are inferred from the relationship between sequences

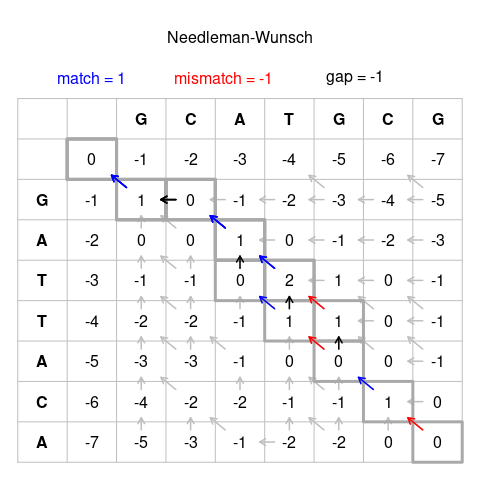
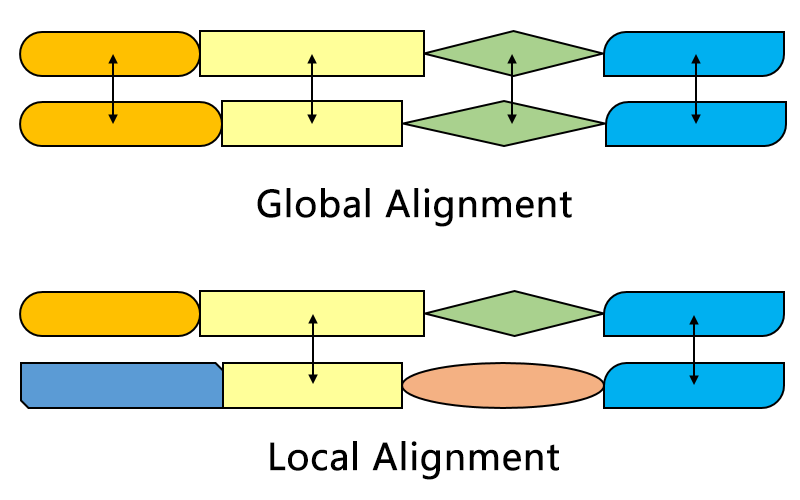
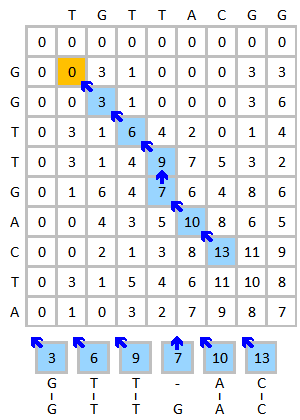
Important biological algorithms are about alignment and search
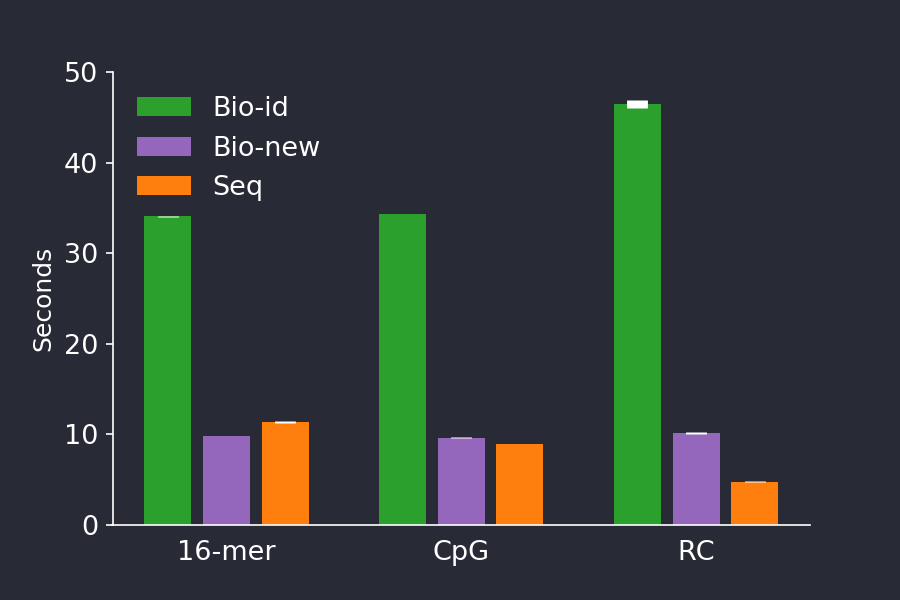
BioSequences.jl competes with special-purpose languages
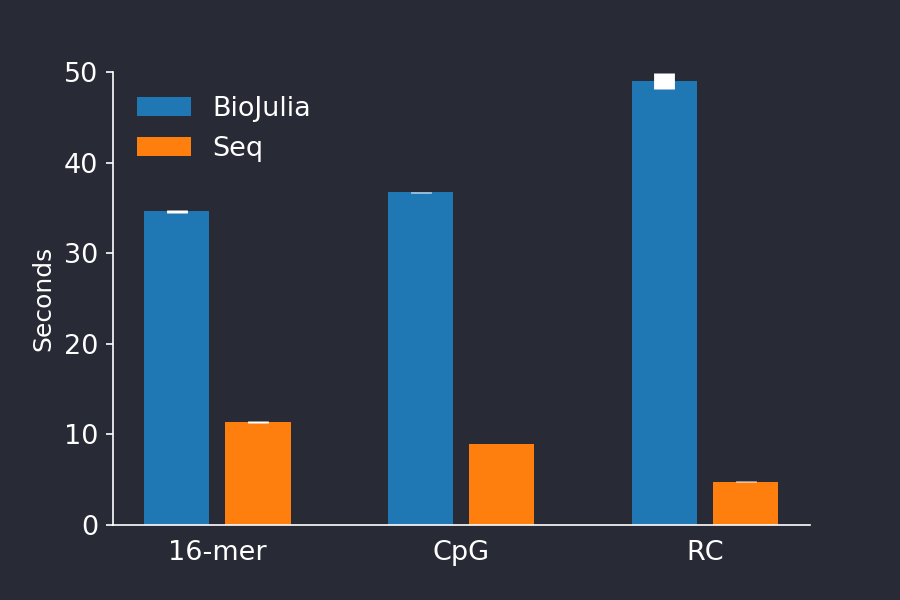
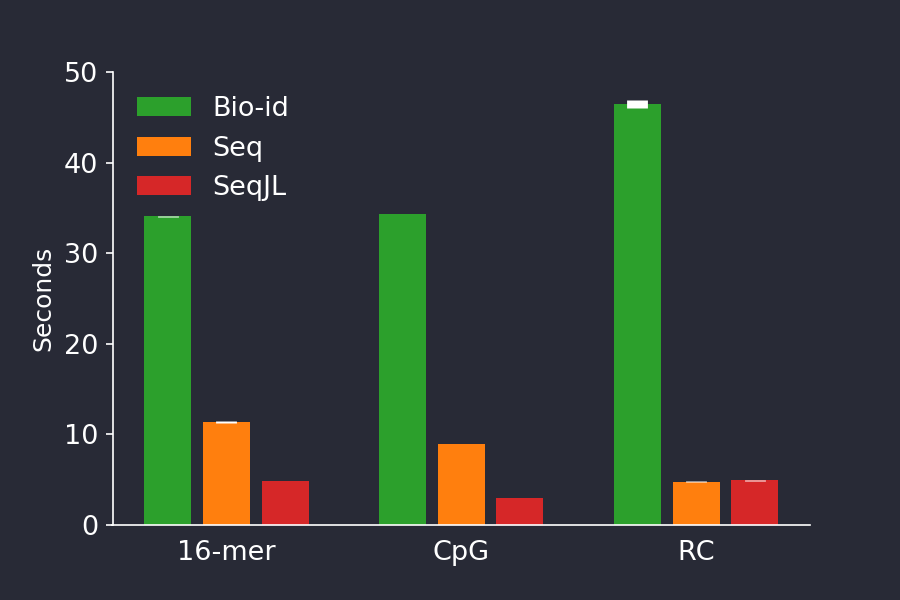
BioSequences.jl competes with special-purpose languages
Automa.jl enables the construction of correct but efficient file parsers
Automa makes Deterministic Finite Automata 
fasta_regex = let
header = re"[a-z]+"
seqline = re"[ACGT]+"
record = '>' * header * '\n' * rep1(seqline * '\n')
rep(record)
endmachine = let
header = onexit!(onenter!(re"[a-z]+", :mark_pos), :header)
seqline = onexit!(onenter!(re"[ACGT]+", :mark_pos), :seqline)
record = onexit!(re">" * header * '\n' * rep1(seqline * '\n'), :record)
compile(rep(record))
end

BioMakie.jl enables easy plotting of protein structure
using BioMakie
using GLMakie
using BioStructures
struc = retrievepdb("2vb1") |> Observable
## or
struc = read("2vb1.pdb", BioStructures.PDB) |> Observable
fig = Figure()
plotstruc!(fig, struc; plottype = :ballandstick, gridposition = (1,1), atomcolors = aquacolors)
plotstruc!(fig, struc; plottype = :covalent, gridposition = (1,2))
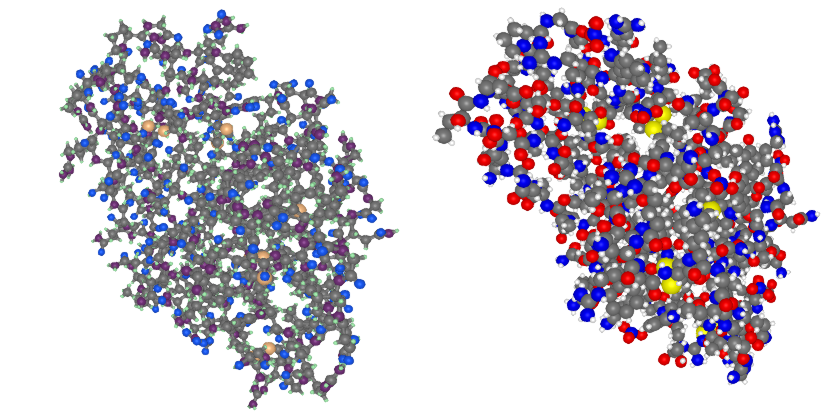
BioMakie.jl enables easy plotting of protein structure
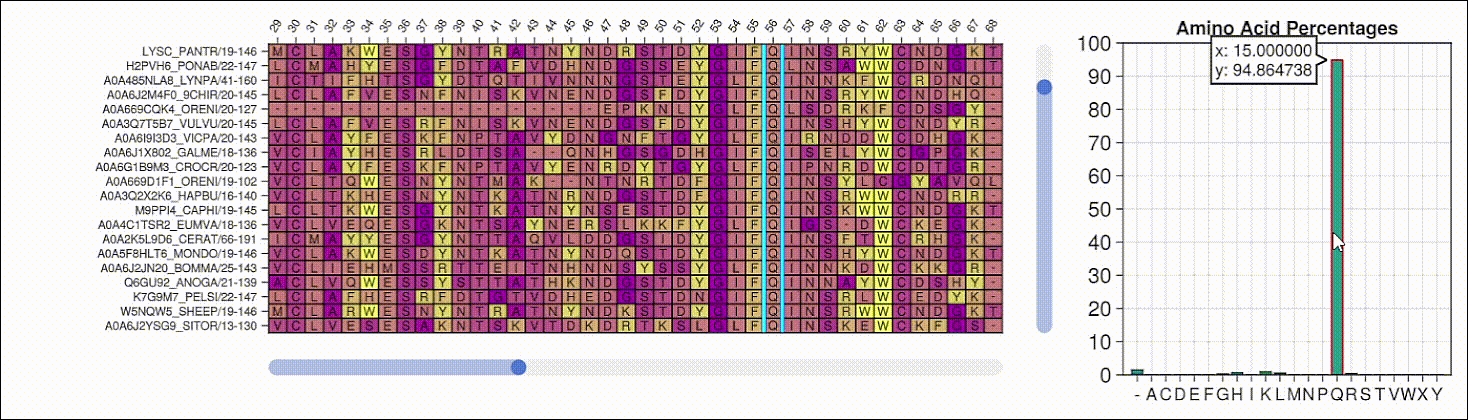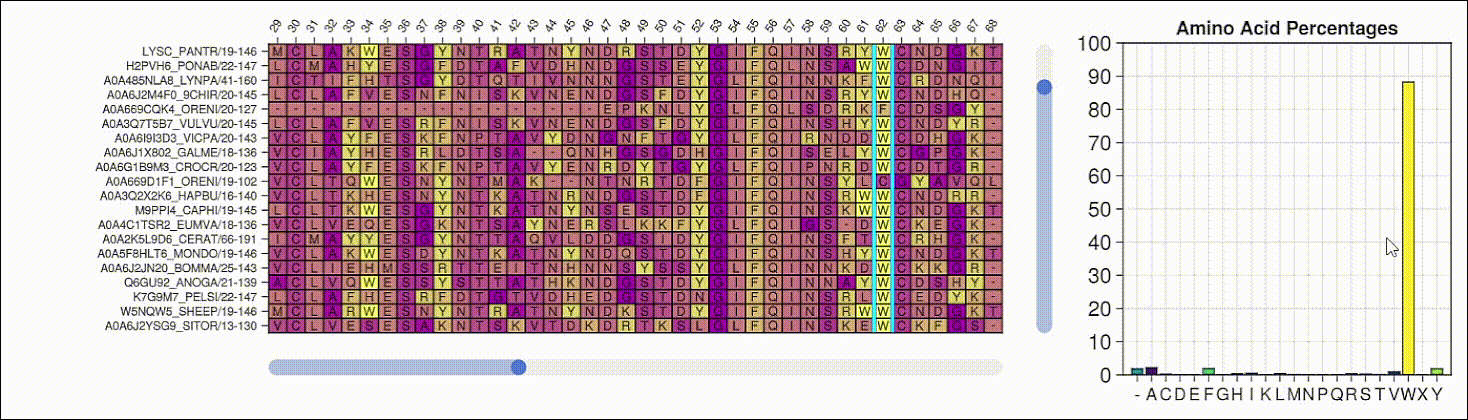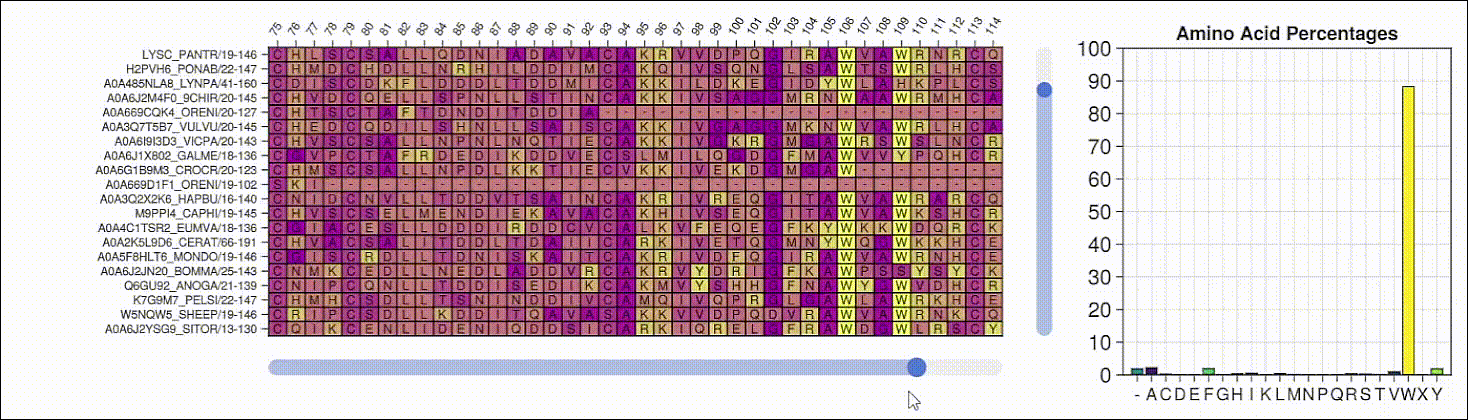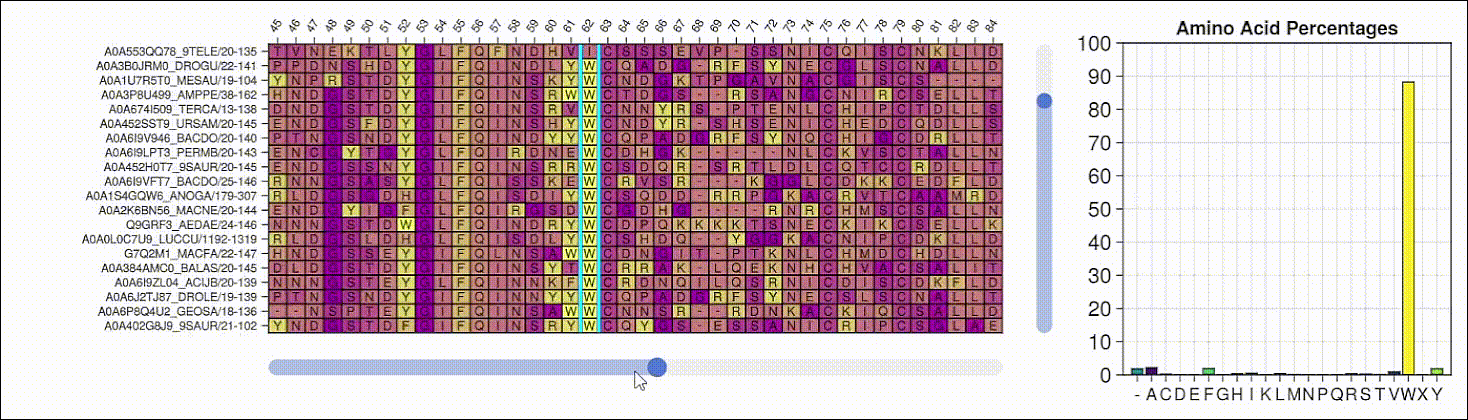

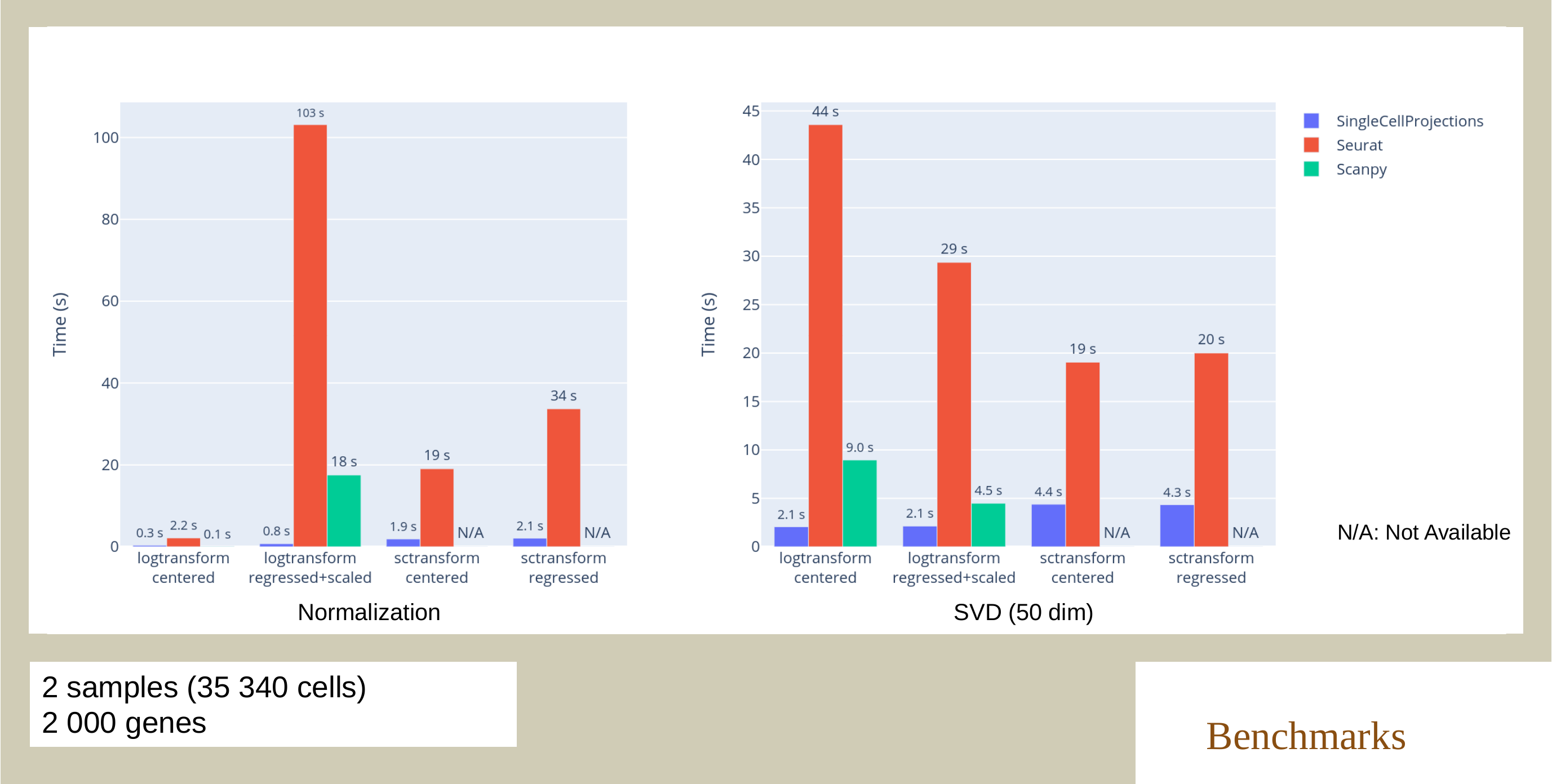
SingleCellProjections.jl enables fast, memory-efficient scRNAseq

SingleCellProjections.jl enables fast, memory-efficient scRNAseq
Pipeline for studying the gut microbiome
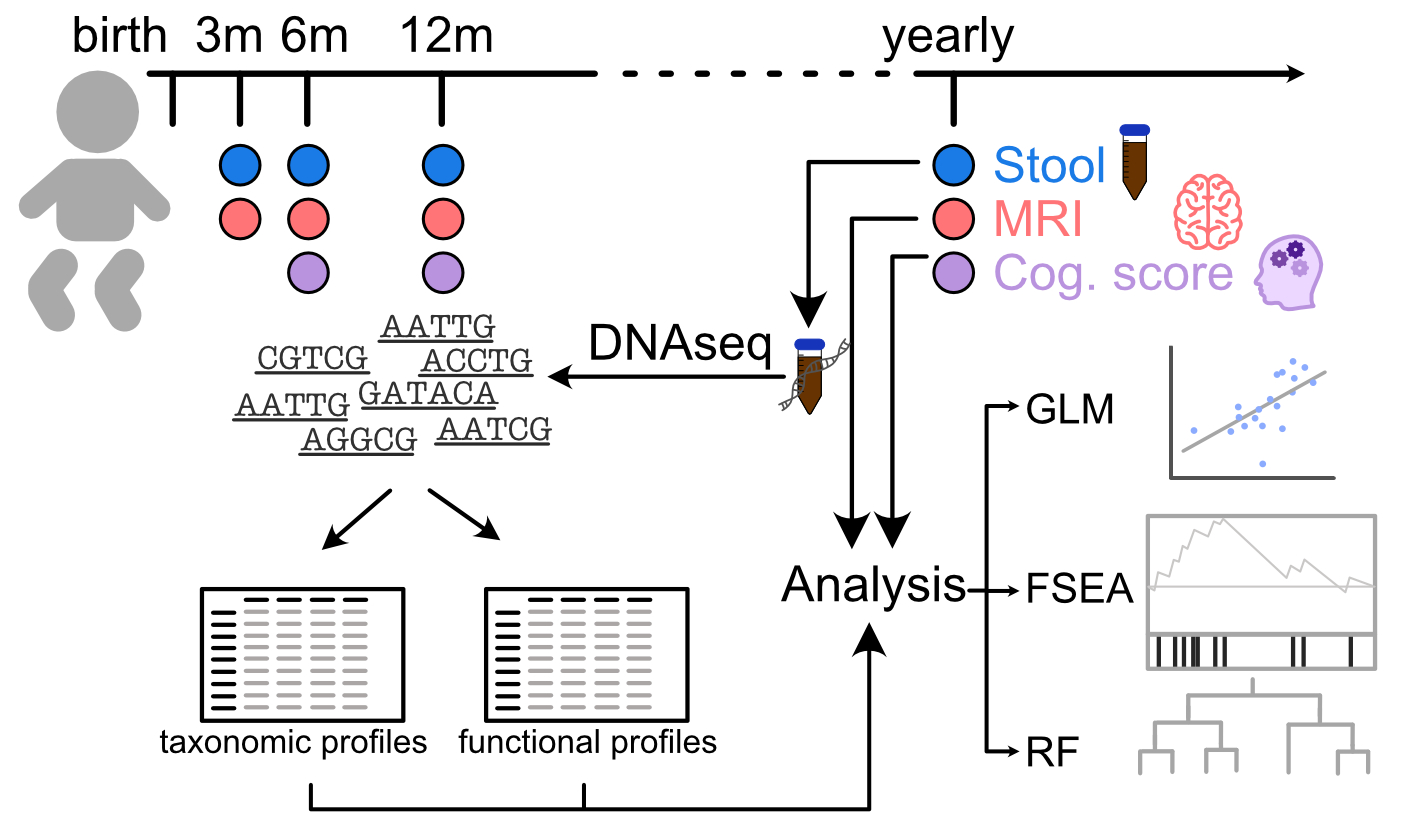
BiobakeryUtils.jl wraps command line tools

- Tools written in python with CLI
- Lots of python and binary dependencies (usually managed with
conda) - Each tool has large associated databases that are not installed along with the tool